
How to Beat Cancer with Viruses: An Interview with Beata Halassy

After stumbling upon her paper, I contacted Beata to interview her. I wrote the previous article alone—although she checked it to make sure I wasn’t saying anything stupid. Today, I share with you an interview we carried out back-and-forth in writing. I wanted to better understand what she went through and the potential of viruses to fight cancers. I also talked with Dr Alan Parker, Head of Solid Cancers & Prof of Virotherapies at Cardiff University, and Dr Stephen Russell, a professor of medicine & hematologist at the Mayo Clinic and founder of a company that explores using viruses to fight cancer. Both were acquainted with Beata’s research. Since this can get technical sometimes, I completely rewrote both the questions and answers to make it understandable by anybody. As a result all words are actually mine, and so is every single mistake. I asked Drs Halassy, Russell, and Parker to cast an eye over the article to make sure it makes sense and reflected their voice.
TOMAS PUEYO: Why would viruses succeed at killing cancers? Is it because the tumor suppresses the immune system, so viruses don’t face opposition inside the tumor and have free rein to attack its cells?
DOCTOR BEATA HALASSY: The main reason why our immune system doesn’t fight against cancer is that cancer cells are our OWN cells. They have simply lost the mechanism to stop proliferating. The immune system1 reacts primarily against foreign substances. That’s the reason why the immune system doesn’t beat cancers.
TP: OK, then why are viruses more likely to kill cancer cells than healthy cells?
DOCTOR STEPHEN RUSSELL: There are two phases to the virus attack on cancers. The first one is when the virus attacks the tumor directly. To understand that, we must first understand how individual cells defend themselves against viruses, without the help of specialized immune cells.
When a virus attacks a cell, it can take over its machinery and start reproducing itself. It can reproduce a lot—sometimes 10k to 100k viruses for every cell they infect!
Cells normally defend themselves from this type of attack in three ways:
Play dead: They stop synthesizing proteins and nucleic acids. This makes the cell environment unwelcome to viruses, and prevents them from taking over a working machinery.
Kill themselves: This is called apoptosis. A cell infected by a virus will simply die before the virus has a chance to make any progeny.
Scream: The infected cell releases interferons, molecules that bind to surrounding cells and effectively inform them that the infection is coming. Alerted, these surrounding cells can now defend themselves better, like playing dead.
Cells will do a combination of these 3, so in order to succeed, viruses must combat these defenses. How do they do that? Besides their genetic material, viruses also carry additional genes that allow them to shut down the cell’s defense mechanism. They prevent the infected cell from playing dead, dying, or screaming. The virus is effectively binding and gagging the cell as it takes it over.
That’s with normal cells. But cancer cells are not normal. They proliferate out of control. How do they do that? They produce a lot of proteins and don’t die. And to do that, they generally eliminate defenses 1 and 2: They can’t play dead or commit suicide anymore. But that’s exactly what the virus wants!
In other words: It’s not that viruses prefer attacking tumor cells. It’s that tumor cells have lost their self-regulation mechanisms, crucial for stopping their own proliferation but also to stop viruses. The cancer cell can’t defend itself from the virus because it can’t play dead or commit suicide. So for each tumor cell that fails to defend itself against a virus, it might produce 10k to 100k viruses that will then attack neighboring cancer cells.
TP: Got it, so the way viruses kill tumors is simply because they don’t defend themselves as well against viruses
Doctor Alan Parker: That’s the first stage. The second stage is that the immune system will also pile on.
Cancers hide well from the immune system because they are cells from the same body, but also because they have some mechanisms to fend it off. For example, tumors can surround themselves with a dense network of cells and proteins. This acts like a physical shield, making it harder for immune cells to penetrate and attack the cancer cells. They can also mask their identity, for example by hiding from their surface the molecules that would identify them,2 or by telling immune cells they are kosher.34
But once a virus penetrates a tumor cell and replicates, it will kill the tumor cell, which will break and release its content into its surroundings.
This massive destruction releases lots of dead cell parts, virus parts, and whole viruses, and the immune system reacts by sending lots of immune cells. They are primarily there to clean up the virus, but since they’re here, they start noticing specific tumor receptors (“neoantigens”) from the pieces of dead tumor cells. Thanks to that, they learn to also identify live cancer cells. Immunity directed to the tumor starts to build up, which might prevent further tumor growth, and can even hopefully act distantly, in other parts of the body.
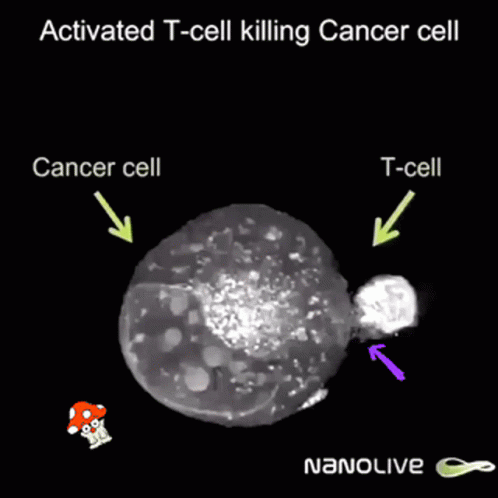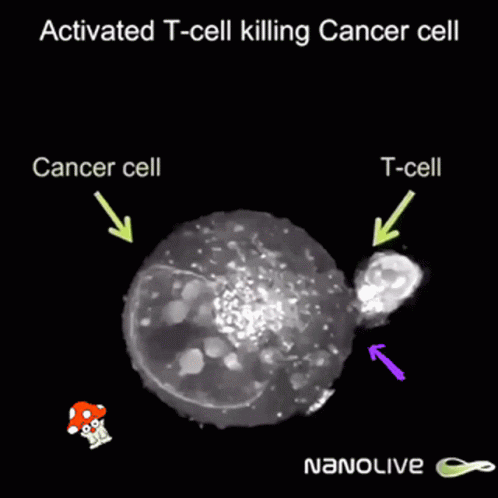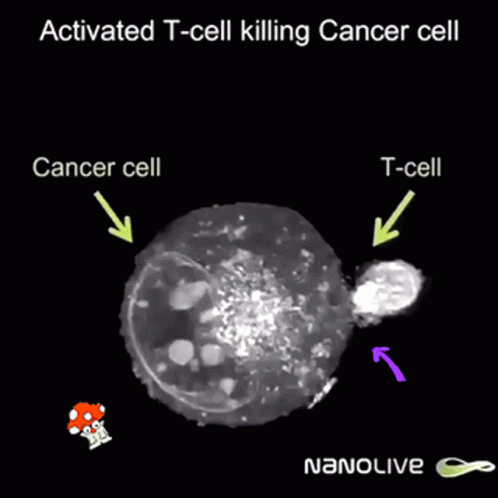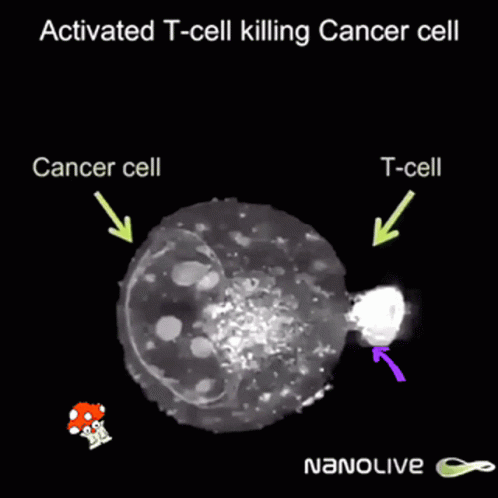
So after the direct action of the virus, antitumor immunity is built due to two facts:
Recruitment of many immune cells within the tumor milieu
Higher availability and visibility of tumor neoantigens
TP: Wow. So to summarize, what’s happening here is:
We inject the virus inside the tumor
Stage 1: Virus Attack
The virus tries to attack normal and cancer cells alike
But the cancer cells are much easier to attack, because they don’t play dead or commit suicide
So the virus will be much better at penetrating cancer cells and killing them
Stage 2: Immune System Recruitment
In the process, it floods the area with viruses and pieces of dead cancer cells
The immune system notices this destruction and sends in tons of immune cells
They clean up the virus and dead cell parts, but in the process they notice this weird neoantigen—pieces of tumor that they didn’t easily notice before, but now they do, and it helps them identify tumor cells
So now the many immune cells start killing tumor cells
Now that they’ve learned about the tumor cells, they can travel around the body and kill more
Doctor Stephen Russell, Doctor Alan Parker: At a high level, yes.
TP: I know past results of oncolytic virotherapy as a last resort have been disappointing, and we hope that bringing it to the first line of treatment will go much better. But if that ends up being true, then Stage 2 should be more important than Stage 1, right? If the viruses were so performant on their own, they should succeed really well in an immunocompromised patient, but that's not what we see from what I can gather.
Doctor Stephen Russell: Yes, immune system recruitment is key to success. No matter how extensively the virus spreads it will never kill more than a fraction of the tumor cells in the patient so we’ll always have to rely on the immune system to mop up. But for full activation of the anticancer immune response, we do need to have the virus infect and kill a sufficient number of tumor cells. But we don’t know what the threshold is for “sufficient” and it probably varies a great deal from one patient to the next…
TP: I do hope we get much more research on this. Do we need special viruses to get all these benefits? Do we need to engineer them? Or are basic viruses enough?
DBH: Viruses can be optimized to fight cancers, and they certainly will in the future, but in this case we didn’t use any engineered virus. It was good enough to use simple viruses that either exist in nature (VSV) or are approved for pediatric use (MeV).
TP: So how did you pick the viruses?
DBH: Normal cells carry receptors, or rather are able to expose some receptors on their surface, which allow other cells to identify them—including the immune system, which reads these receptors to recognize the cells as being part of the body.
Viruses have evolved to learn how to attach to some of them and use them to enter the cells. Frequently, they can latch onto more than one of these molecules.
Now, as we discussed, tumor cells and normal cells are nearly identical, with the major difference that tumor cells don’t stop replicating. So tumor cells also have these receptors that viruses can hijack.
We wanted viruses that could easily identify the receptors in my tumor. Breasts have lots of epithelial cells—cells that line up our vessels and ducts, for example milk channels, blood vessels, intestines, glandular ducts.
Both of the viruses we chose—measles and VSV—are great at identifying epithelial cells, so that’s why we chose them.
TP: That makes sense. Any other reason why you went for these ones?
DBH:
It had to be a virus that infects epithelial cells. Both VSV and MeV (Measles Virus) do infect such cells in culture.
It had to be a virus that someone had tested and for which they had demonstrated potential against breast cancer. This was true for MeV clinically, and VSV preclinically.
It had to be a virus we had in the laboratory and which we were extremely skilled at handling.
It had to be a virus that can’t cause serious illnesses in humans.
It had to be an already existing virus in the environment. We did not want to risk that some genetically modified virus spreads from patient to someone in an uncontrolled fashion.
Measles and VSV both fit the bill.
TP: Thank you. Super clear. I’m no expert in this, but it sounds like a very promising avenue to fight cancer, yet we haven’t had much success testing it since the 1940s.
DBH: Back then, we knew very little about virology. The field was in its infancy. Now we have so many more tools! We can figure out the genetic code of viruses, we can edit it, we can suppress their aggressiveness, we can select the best strains, we understand how viruses work a a molecular level, how cells react to them, how the immune system intervenes… What didn’t work then can work now.
TP: So that’s why there are so many ongoing randomly controlled trials (RCTs) testing this approach with humans. That makes sense. But then why is this field progressing so slowly?
DBH: When this approach was tested on mice, results were very promising. But then we did lots of RCTs on humans, and results were far below expectations. So the conundrum was: Why on Earth are we not getting similarly good results in humans? Is it just a difference between mice models and humans, or is there a unique thing we’re doing differently between mice and humans that might explain this difference? We hypothesize it might be both.
Normally, when there’s a new potential treatment for cancer, the current approach is to start by testing it as a last resort: We first attempt what’s been tried and proven to work, like surgery, chemotherapy, biological therapy, or radiation. Only when those options fail do we explore experimental treatments, including oncolytic virotherapy. This means it’s initially used in patients with advanced or resistant cancers.
For most novel cancer treatments, this slows down progress because they’re facing the most challenging cases. But in the case of oncolytic virotherapy, this is especially bad, because we need to recruit the immune system to do a big chunk of the work, but by the time the cancer is advanced, the immune system is extremely weak due to both the cancer and the treatments against it.
The consequence is that oncolytic virotherapy looks like a much worse approach in many of these RCTs than it would probably be if we tested it as a first line of treatment rather than a last resort.
TP: So you think clinical trials with oncolytic virotherapy should be done earlier in the cancer process.
DBH: Yes, they will probably work much better than they do today, and maybe they don’t always work as well as other treatments, but they are certainly less destructive. So maybe we can use them as first line or treatment, or in combination with other treatments.
This is why I cared so much about publishing my case. It may encourage the researchers already working on these drugs to target earlier stages of the cancer in clinical trials, as a neoadjuvant treatment [=pre-treatment before the main intervention] instead of conventional approaches like chemotherapy with/without immunotherapy.
If we could get it as a first line of cancer treatment that is not toxic to the immune system and health of the patient, this would be an amazing achievement. It may also promote the development of combined treatments, with more than just one oncolytic virotherapy. It also opens the possibility that some already existing viral strains that have been safely used over the years for prophylactic vaccination against viral diseases could be used as oncolytics, if formulated and applied differently. In other words—some already approved products might be studied and developed for novel application. I hope that the article will stimulate investigations in all these directions, and that will finally provide novel, less toxic, and at least equally effective—if not better—therapies to cancer patients.
TP: There you go, readers. If you work in cancer research, or you know somebody who might, or you know somebody with cancer who might be a good candidate for this, or know somebody in pharma or regulators working on cancer, it might be useful to explain this approach to them. Beata, are there other causes of slow progress with oncolytic virotherapy?
DBH: Yes, we face many other challenges. The main ones I can think of are:
Therapies require a huge amount of viruses, which are challenging to produce and store without losing virus infectivity.
There are also safety concerns: The majority of viruses are genetically modified organisms, which, as replicative entities, pose a potential threat if they pass from person to person.
Also, researchers often use cell lines that originate in cancers, so regulations are very rigid, like the requirements of purification steps to remove host cell proteins and nucleic acids from viruses.
Doctor Alan Parker: One of the things that I found fascinating is the approach that Dr Halassy took to fight the virus. Choosing two viruses instead of one, and making 11 injections in total… It sounds like she picked a great approach based on her deep expertise in the field, otherwise there’s no way anything like that would have been approved for an RCT. Even with this evidence, I wonder how we would get something like that approved. I wish we did.
TP: Yes, we should take our best guess when designing trials!
Doctor Alan Parker: I also don’t know how to finance RCTs to make this happen. Pharma companies have an incentive to test a single vaccine that they’ve patented, but Beata tested two viruses, and neither are patentable.
TP: Maybe they can slightly modify the viruses to make them better adapted to kill the virus. Or maybe governments or nonprofits should pay for RCTs of non-patentable treatments that could nevertheless save lots of lives?
Anyway, let’s now turn to your own experience with this treatment, Beata. Your cancer had come back twice. Is this why you decided on an unconventional treatment?
DBH: The first two times, the cancer had been triple-negative, which is especially nasty.5 We were all expecting that the same type had come back. Triple Negative Breast Cancer (TNBC) is the worst form of breast cancer, there is no specific cure for this type, and if it returns, it returns quickly which brings poor overall prognosis (“likely course of a medical condition”).
But this second recurrence was found for the first time to express the receptor for human growth factor (HER2). It is known that cancer can modify its phenotype, but also that one cancer can be the mixture of several types. My personal belief about what happened here is: The first recurrence in 2018 was TNBC and was surgically excised. However, after the surgery the seroma remained at the site of excision (“a pouch of fluid remained where we cut the cancer”). This entrapped body fluid perhaps fed the remaining cancerous cells. In such a supportive environment, it may have expressed receptors for human growth factor, whose activation additionally stimulated the growth of the cells. It was a fast growing tumor.
TP: After the treatment with the measles virus and VSV, you got the tumor excised. Then, you treated yourself again with some measles a couple of months later. Why inject more virus? Why around that time? Why measles and not another virus?
DBH: There are no protocols for any of this. I wasn’t trying to apply any known rule, but rather try to guess, based on what I know, what is the most likely treatment I can get to maximize my chances that the cancer won’t come back.
When we cut my tumor, maybe some cancer cells were still present (a single one could be enough). They can easily escape such a surgery, so we decided to inject the viruses one more time, to catch any cancer cell that had escaped. So that’s why a new injection.
Why measles? When treating a cancer with viruses, you want to give them time to attack the cancer, so the immune system can’t be too well trained on your virus. We had just used VSV, so my immune system was probably very attuned to it. VSV also has a problem, which is that it had never been used intentionally in humans before.6
We’re very comfortable with the measles virus: We know it very well because we’ve used it for years in humans for children’s vaccination. Also, a longer time had passed from the previous injections, so we supposed that the immune response against this virus had already waned a bit, which hopefully meant that the virus would have more freedom to operate.
TP: Your team excised the tumor after just two months (9 weeks) of treatment with the virus. By that time, your measures suggest the tumor had shrunk by 63%. How fast is that?
DBH: According to my oncologist, existing neoadjuvant (=“therapy before surgery”) therapy in HER2 positive tumors lasts from 18 to 27 weeks, depending on the type of the protocol. Such protocols generate a complete pathological response7 in 50-60% of patients. For TNBC, existing protocols last on average 20 weeks, and also generate a complete pathological response in 50-60% of patients.
TP: The paper mentions the tumor was very defined and hard. So why not just excise it? Why treat it first with viruses?
DBH: For fear of recurrence. This was the third time the cancer had appeared. But for the first time, it was expanding to neighboring tissues—skin and muscle—meaning it had demonstrated metastasizing properties. So the clinical picture was worse than in the previous two encounters with the tumor.
Also, neoadjuvant therapy using conventional chemotherapeutics to shrink the tumor before the surgery has already been proven to have better outcomes over conventional therapy, which starts with excision.
TP: Your paper has become quite popular, with tens of thousands of reads. What do you think of that?
DBH: I’m very glad that so many people have been discussing the paper, but I think it’s for the wrong reasons. I think this case indicates that oncolytic virotherapy is very promising as a first line of treatment, but unfortunately most of the debate has been around ethics.
TP: Why do you think the ethics have been so polemic?
DBH: The question of the ethics of self-experimentation had been consistently raised while we were trying to publish this article, specifically with the issue of trying to do this without an ethics board. As it was a case of self-experimentation, I did not consider it necessary to ask for anyone’s approval. In addition, all this was happening during the COVID-19 epidemic, when all medical efforts were involved in treating COVID-19 infected people. I do not think that anyone would have been able or interested in providing ethical approval for something so unconventional, at such an unconventional time.
Given the hesitance to publish the paper, I searched the history of self-experimentation (a nice review is available by Hanley et al.) and the history of ethical principles in science generation, and learned that there is indeed nothing unethical or illegal in self-experimentation.
TP: That was the main concern, but were there others to note?
DBH: The only big other concern was about the use of laboratory-grade viruses instead of clinical-grade. The difference is that clinical-grade is purer and better characterized, and the content is quantified and defined with more reliability, which means we’re very confident that it was the virus that shrunk the cancer, not something else inside of the lab-grade injection.
Clinical grade is clearly better, but that wasn’t available to me. So the question becomes: Is laboratory-grade good enough to prove it was the virus that did the work?
I think it is, because otherwise, if we follow the argument that lab-grade is not good enough, most experiments in mice would never get published: They use lab-grade preparations too.
The question is rather: How pure was our lab-grade virus? And that’s where it matters who our lab is. We have a huge amount of expertise in virus preparation characterization. We have been granted patents on virus purification; we have publications on virus preparations during COVID-19 time. I am very confident that these preparations were quite well characterized, particularly the quantity of infective virus.
I think in this type of paper, what matters is that we clearly disclose the entire process: How viruses were prepared, how they were characterized, what is the level of our expertise… That way, anyone reading the article will be fully informed, which is the point of a paper.
TP: It took many attempts to get this published: 13 rejections in 2.5 years. Has this been your experience in the past?
DBH: I have never experienced such a level of paper rejection in my whole career with close to 80 publications published in international journals.
We did expect it to be harder than other papers we had published for a couple of reasons. First, we aimed at even more prestigious journals than usual given the uniqueness and novelty of the case. Second, our team has lots of virology and immunotherapy expertise, but no oncological expertise. So we expected some pushback, but not at this level.
In several journals, the paper was immediately rejected due to ethical concerns. We even received three positive reviews in one of the journals, and then a completely unexpected rejection by the academic editor.
From another perspective, this might have been for the best. This has allowed for time to pass. Now I’ve been cancer-free for 4 years, which is much stronger support for this approach.
TP: Well, I’m glad your team and you finally got the paper published. And more importantly, that you’re alive and cancer free! I hope many people hear about your case, and we do our small part to help cure cancer.
DBH: Thank you!
Through its innate and adaptive branches.
Normally, cancer cells produce abnormal proteins called neoantigens that the immune system can recognize. Some cancer cells can reduce the production of these antigens or hide them, making it harder for immune cells to detect the tumor as a threat.
Tumors can produce proteins, such as PD-L1, that bind to "checkpoints" on immune cells—molecules they use to identify foreign bodies. When these checkpoints are activated, the immune cells stop attacking, essentially telling the immune system, “Don’t attack me; I’m part of the body.” In fact, some drugs fight this by binding to the tumors’ proteins, which then can’t stop immune cells. One way to imagine this is if immune cells are police and they carry around locks, asking cells to present the key that unlocks them. This type of drugs is made of other locks that fit into the virus’s keys, and so they’re left without keys to unlock the police (immune cells)’s locks.
Tumor cells can also release chemicals (like cytokines) that suppress immune cells, especially T-cells, which are crucial for identifying and killing abnormal cells. This tricks the immune system into being less aggressive against the tumor.
As we saw in the first article, a triple-negative breast cancer is called so because it doesn’t have enough estrogen receptors, progesterone receptors, or human epidermal growth factor receptors—the three receptors that can be targeted by approved anti-cancer drugs traditionally used to treat it. These cancers are the deadliest form of breast cancer, hard to treat, with the worst prognosis and overall survival.
There are some concerns that VSV can be neurotoxic if it reaches the brain.
Pathological complete response (pCR) is defined as the absence of residual invasive cancer on hematoxylin and eosin evaluation of the complete resected breast specimen and all sampled regional lymph nodes following completion of neoadjuvant systemic therapy.
Curiously enough, now news are spreding about using ivermectin and fenbendazole to cure cáncer allegedly because it promotes apoptosis of malignant tumour cells. Further research is needed but even Mel Gibson has reported on Joe Rogan's some miraculous cases of people overcoming last stages of fatal cancer by these means.
Really interesting topic and interview.
Thanks for the good summary and making it understandable, Tomas.
It would be good to have a way to volunteer for this type of study. Ethics seem to have been taken to the extreme in medicine these days (I understand the pros of this) but I like the analogy from the last article about the car industry.
And I think the pharmaceutical industry have a little too much invested in treating vs curing illnesses.
I’ll be watching this topic with interest.